Chapter 5 Discussion
The aim of this thesis was to characterize undetermined sequences from metagenomic sequencing, focusing on a dataset of throat swab samples. This aim should not be confused with just trying to classify as many sequencing reads as possible. Instead, it was also deemed important to gain insights and possibly understand some of the relationships between the different variables.
This work should be the basis for further investigations and potential optimization approaches of the workflow and analysis pipeline if required.
We showed that the classification success correlates with the sequencing read quality (Q30 rate and read length). Lower Q30 rates and shorter read lengths were observed in unclassified reads when compared to the classified ones.
If the primary aim was to minimize the fraction of unclassified reads after comparing them to a set of reference sequence databases, one could make the quality filtering steps more stringent. In my opinion, for the application of the study, this might not be the right choice. When applying metagenomic sequencing for diagnostic purposes, the main aim is to make the test as sensitive as possible, especially since this method is currently usually less sensitive when compared to a specific test (e.g. quantitative PCR).56 Removing more of the lower quality reads might lead to a decrease in sensitivity in favor of less unclassifiable reads, which is not a good trade-off.
The only drawback of not removing lower quality reads at the beginning of the bioinformatic pipeline is, apart from a bit slower processing speed, that these low-quality reads could lead to a false-positive result. However, there are other measures to avoid false positives, which are in place anyway because it is an important topic when evaluating metagenomic sequencing results for clinical applications.
A linear regression model was fitted to the data to predict the number of unclassified sequencing reads given a number of filtered reads. Apart from some outliers, the proposed model fits the data relatively well and has the advantage of interpretability. The intercept represents the fixed number of sequences that cannot be classified by VirMet and is the same for all samples as they are prepared and sequenced using the same protocol. The slope describes the estimated additional unclassified sequencing reads with the increasing number of filtered reads.
The outliers which did not fit that regression well (mainly coming from the DNA workflow) have a higher than expected number of unclassified reads. The residuals of these samples, which is the difference between the observed number of unclassified reads and the predicted number, can be used to automatically detect potentially interesting samples. One can either use the residual values themselves as a quantitative value or, as it was done in this work, define a certain threshold that, when exceeded, flags the sample. The threshold applied here is relatively high with 500’000 reads and would likely not be useful to detect novel organisms of which there is no sufficient amount of genetic material (DNA or RNA) present in the sample. In a clinical setting, it is therefore probably better not only to fixate on the residual values but also to consider the patient’s symptoms and whether or not a causative pathogen could be found.
To prove that this simple method is suited to flag potentially interesting samples, the residuals were compared with the Q30 rate differences (\(\Delta\)Q30 rate) between classified and unclassified reads of the same sample. Because not all samples followed the overall pattern described in the previous section (unclassified reads have a lower quality), it was hypothesized that a low \(\Delta\)Q30 rate indicates the presence of an undetected organism. The correlation which is observable between the residuals and the Q30 rate difference shows that the results of these two different methods largely overlap when it comes to flagging potentially interesting samples.
If the quality of a sequencing read is fine and yet it cannot be classified, the choice of reference database used is likely the cause of the inability to classify it. Only reads of which a genetically not too distant related sequence is present in the used reference database can actually be classified. It is therefore important to use reference sequences that cover as much of the genetic diversity as possible.
But since a complete set of such reference sequences of all organisms is not yet available (even the human genome is only just now about to be complete57), alternative unsupervised or semi-supervised methods have to be applied to make up for the incompleteness of the databases.
Applying a semi-supervised approach by which de novo assembled contigs are combined with the VirMet class labels helped classify additional sequencing reads which were previously unclassified. While on average only a small fraction (10.8%) of previously unclassified DNA reads could be classified, more than half (67.7%) of the previously unclassified RNA reads could be classified. The additionally labeled reads originate from all classes which suggests that there is no specific bias towards one of the classes but all are more or less affected. This does not make finding a solution easier but underlines that more exhaustive reference databases of all classes would likely help to reduce the fraction of unclassified reads.
Focusing on the samples flagged using the linear regression approach it is notable that they have mostly a small proportion of “unclassified not in contig” reads and a relatively high one of “unclassified in contig”. This indicates once more that the method used for flagging these samples might be appropriate because it seems that there were contigs formed but they could not be classified. This is what is expected when there is a large number of reads from an organism that is not part of the reference databases present. This pattern cannot be observed within the RNA samples and might be due to the fact that there are fewer unclassified reads remaining after the semi-supervised approach which in turn suggests that the reference databases used are more complete for RNA sequences.
5.1 Open questions
While all these findings partly explain the characteristics of unclassified metagenomic sequencing reads, they lead to further questions that should be addressed in future work.
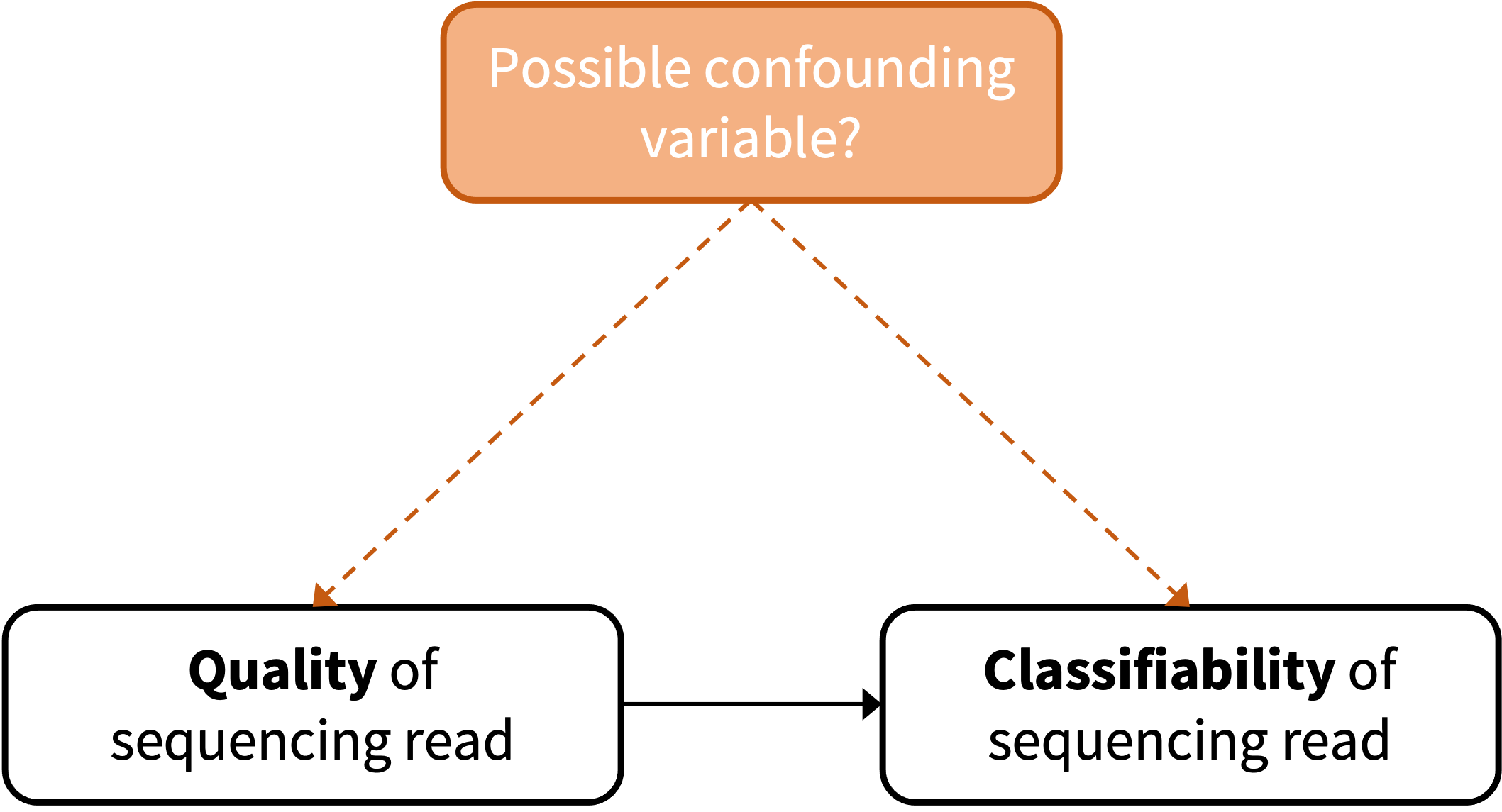
Figure 5.1: Schematic visualization of the association between quality and classifiability, and a possible confounding variable.
An important uncertainty to be resolved is whether sequence read quality and classifiability are directly associated or if there is a confounding variable involved, as shown in Figure 5.1. This could be investigated by simulating sequencing reads and comparing the classifiability as visualised in Figure 5.2.
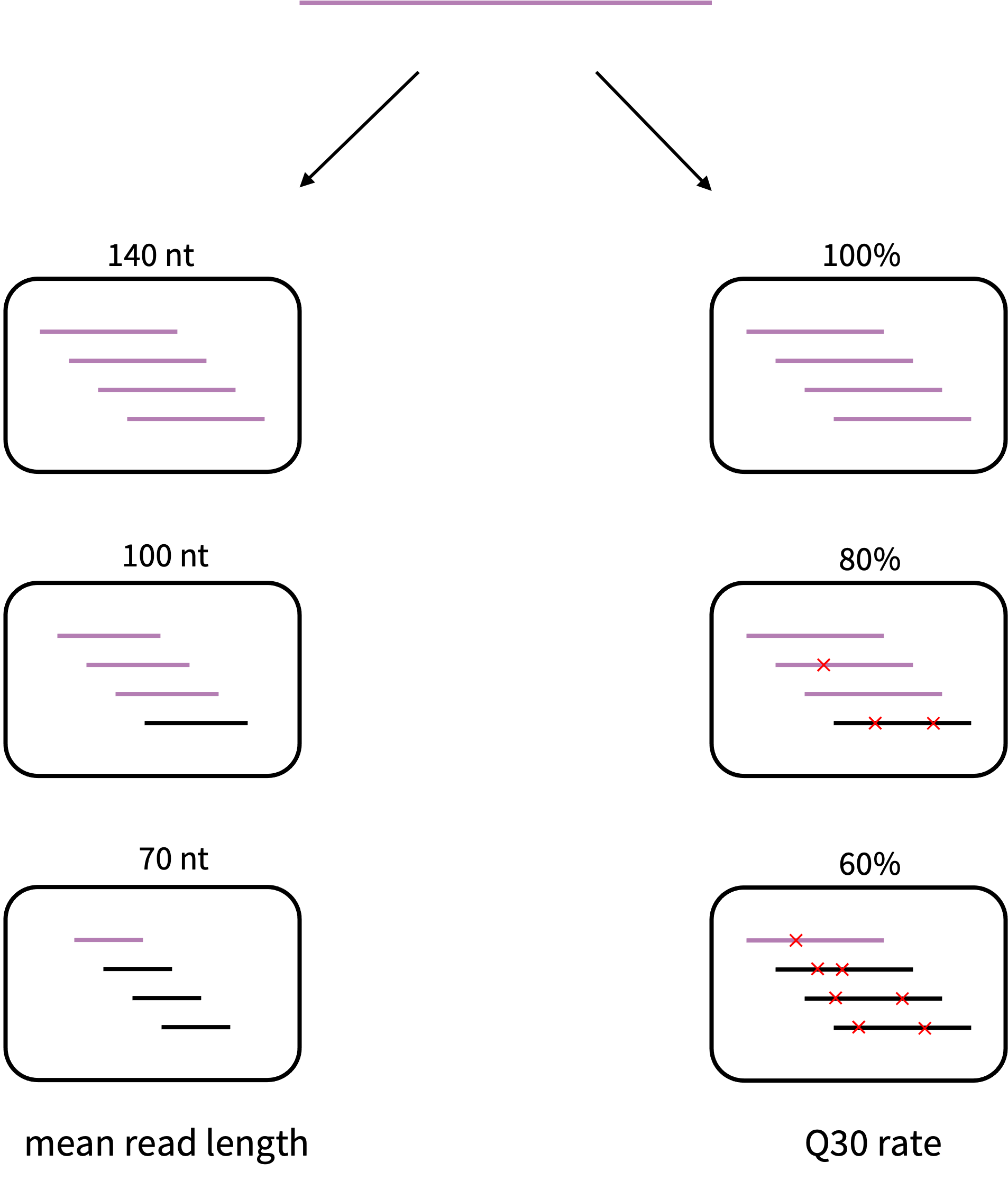
Figure 5.2: Proposed simulation experiment to investigate the influence of sequence read quality on classifiability. Starting from a known genome from the reference databases, one could simulate datasets (black boxes) with different quality properties. Black colored reads represent the ones which could not be classified. Red crosses represent sequencing errors.
To further find common patterns between different samples, a cross-assembly could be done. This would involve using all sequencing reads combined for one large metagenome assembly instead of treating each sample separately as it was done here. By then mapping the reads of every single sample back to the assembled contigs, one could detect contigs that have reads present in multiple different samples. This could mean one of the following two things. Either the reads are coming from a common organism (which multiple patients share) or there is a common contaminant introduced by reagents used for sequencing. Both of these options would be interesting and as has been shown, metagenome assembly as applied here is possible and thus laid the basis for further experiments involving something in this direction. The drawback of this approach is that it would use large amounts of memory (RAM) while processing all reads combined. This is the reason (along with time constraints) it could not be included in this work.
If required, the semi-supervised approach could be adapted to return more detailed information. Instead of the very broad classes (human, bacterial, fungal and viral), it would also be possible to list the results in more detail (further up the taxonomic rank) and for example, break down the viral species which were additionally classified. This was not done in this work for simplicity reasons but would be possible in future work.
Lastly, this study focused on the results from metagenomic sequencing of throat swab samples. There are however other sample types processed and sequenced using the same protocol. It would be interesting, now that we have a first impression of what to expect, how these results look like when applied to samples from different sample types (e.g. blood or stool).
References
56. Ebinger, A., Fischer, S. & Höper, D. A theoretical and generalized approach for the assessment of the sample-specific limit of detection for clinical metagenomics. Computational and Structural Biotechnology Journal 19, 732–742 (2021).
57. Nurk, S. et al. The complete sequence of a human genome. bioRxiv 2021.05.26.445798 (2021) doi:10.1101/2021.05.26.445798.