Chapter 1 Introduction
Metagenomic sequencing, a method where non-specifically DNA and/or RNA molecules which are present in a sample is sequenced, can be used as an untargeted approach to gathering information about the taxonomic composition of all genetic material present. Using this method, microorganisms such as Bacteria, Viruses, Fungi, Protozoa, Algae and Archaea can be detected in various sample types.1
In addition to conventional specific diagnostic tests to detect infectious disease-causing organisms in humans, metagenomic sequencing recently gained importance in clinical settings. The advantage of having DNA/RNA sequences as a test result is that it can not only tell if an organism is present but also additional conclusions like drug resistance, spread and evolution of the pathogen can be made if enough sequencing reads can be obtained.2–4
Such a test system, currently restricted to the detection of viruses, was successfully introduced at the Institute of Medical Virology of the University of Zurich. This thesis addresses some of the points which are aimed to be improved.5
During the process of shotgun metagenomic sequencing, a large number of relatively short sequences are generated. It is a major bioinformatic challenge to accurately and efficiently classify these sequencing reads and produce a summary of the composition of microorganism genomes present in a specimen. Especially difficult is the fact that there is a mix of genomes from many different kinds of organisms.
The current bioinformatic pipeline which is in use at the Institute of Medical Virology is called VirMet (github.com/medvir/VirMet). Briefly, raw sequencing reads are filtered using multiple requirements (length, entropy and quality). The remaining reads are then mapped against different databases (human-, bacterial-, fungal-, and bovine-genome). The mapped reads are currently discarded in order to speed up the BLAST6 process where the remaining reads are blasted against a viral-genome reference database. After those steps, there always remains a fraction of reads which could not be classified (Figure 1.1). Those reads, potentially harboring important information, are currently not further regarded in our lab.
Since the reference sequence databases used are far from complete, novel genomes might potentially go undetected. However, it does not necessarily have to be like this. There are approaches that have allowed a de novo assembly of genomes from previously uncharacterized organisms using metagenomic sequencing, such as crAssphage7, Redondoviridae8 or SARS-CoV-29.
Fast detection and assembly of the SARS-CoV-2 genome was crucial to develop specific molecular diagnostic tests and proved the importance of appropriate metagenomic methods which made this possible.10
In this work, the focus is on finding correlations and possible explanations for the unclassified sequencing reads as well as suggesting alternative approaches to overcome this issue.
The dataset used in this project consists of results from 283 throat swabs which were processed and sequenced with the established protocol (github.com/medvir/virome-protocols v2.2.1). Since this protocol splits each sample into a DNA and RNA workflow, there are two sequence read files for each sample which can be analyzed separately.
The throat swabs were collected and analyzed within the scope of a trial where general practitioners could send in samples from patients with respiratory symptoms.
The hypothesis is that most of the unclassified reads are not novel (human infecting) viruses but rather sequencing artifacts and unknown bacteria. This will be tested by applying different methods used for metagenome assembly and through strict quality control/trimming of sequencing reads.
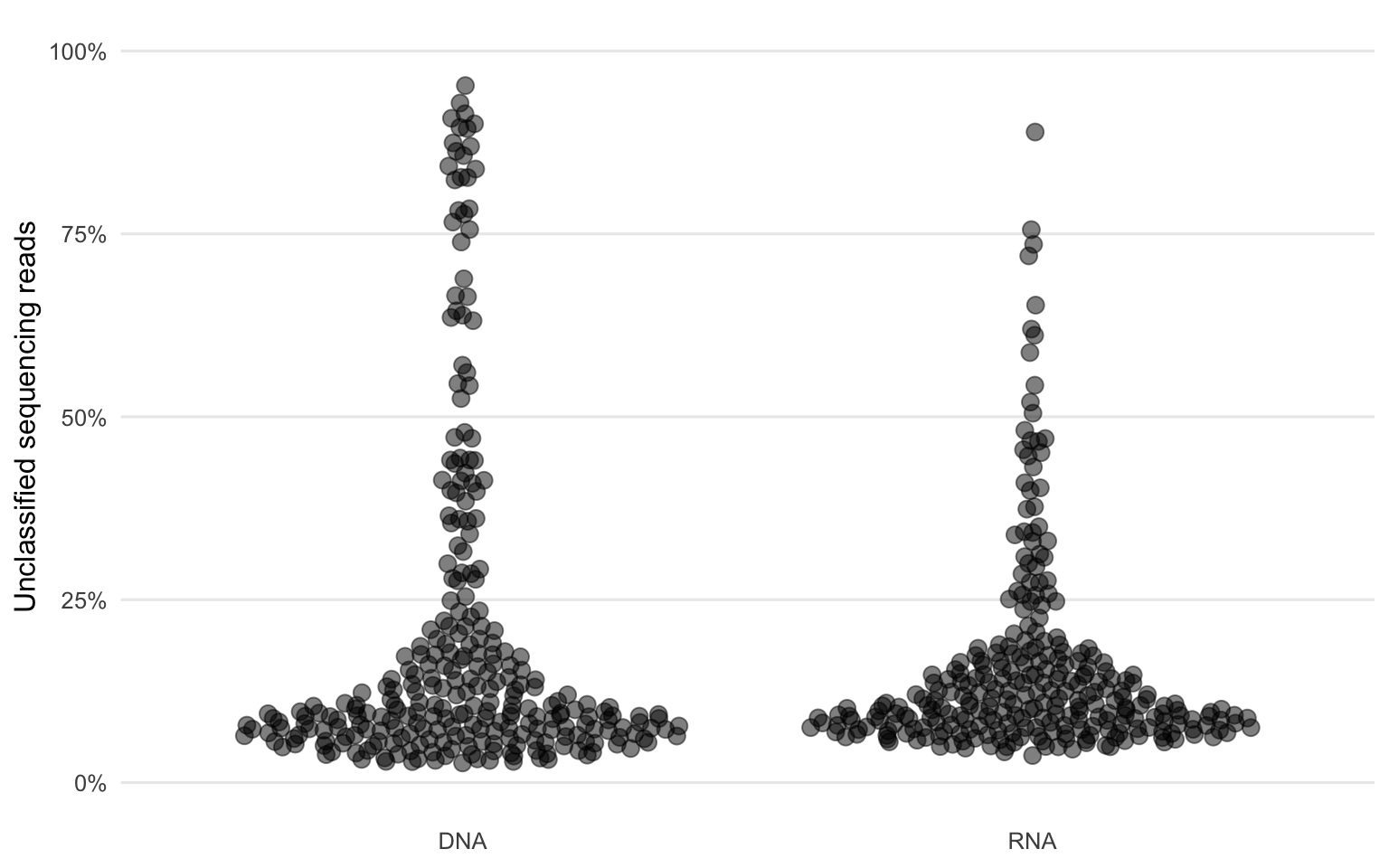
Figure 1.1: Fractions of unclassified reads of all quality-filtered reads demonstrate that in some samples the majority of the sequencing reads could not be classified.
References
1. Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nature Biotechnology 35, 833–844 (2017).
2. Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nature Reviews Genetics 20, 341–355 (2019).
4. Hadfield, J. et al. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
5. Kufner, V. et al. Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes 10, 661 (2019).
6. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990).
7. Dutilh, B. E. et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nature Communications 5, (2014).
8. Abbas, A. A. et al. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract that are associated with periodontitis and critical illness. Cell host & microbe 25, 719–729.e4 (2019).
9. Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).