Chapter 2 Literature review
Ending up with an unclassified sequencing read from shotgun metagenomic sequencing can have different reasons. Therefore there also exist different strategies which can be applied to tackle and study this issue.
In this Section, two such methods are reviewed. First, the topic of sequencing quality and artefacts is discussed, followed by the description of unsupervised and semi-supervised approaches.
2.1 Sequence read quality
All sequencing technologies have a certain error rate and error profile. Factors affecting the read quality, and therefore the possible inability to classify a read, are not just single nucleotide errors but also long stretches of low-quality bases and the presence of synthetic adapters used for sequence library preparation.11,12
The accuracy of each sequenced base is typically measured by Phred quality score (Q score) and added to the output sequences (fastq format).13 The Q score is calculated by using the probability of an incorrectly called base \(P\) as shown in equation (2.1).14
For Illumina sequencing reads the Q30 rate is often used. This number stands for the fraction of the sequenced bases of a sample that have a Q score higher than 30 (base call accuracy of at least 99.9%).
\[\begin{equation} Q = −10 \cdot \log_{10}(P) \tag{2.1} \end{equation}\]
If our goal is to reduce the fraction of sequencing reads from metagenomic sequencing which remain unclassified it’s important to incorporate suitable quality filtering steps in the bioinformatic pipelines. There is no single ideal way or set of parameters we can use to clean all sequencing datasets since those depend on multiple factors such as the source of DNA/RNA, library preparation protocol, sequencing method and platform. Experience and exploratory data analysis of the quality of a data set might help to find optimal parameters. There are, however, also methods that require less prior knowledge. One example is the automatic removal of synthetic adapters by considering k-mer frequencies at the ends of the sequencing reads.15
2.2 Unsupervised and semi-supervised approaches
A straightforward way to classify sequencing reads from metagenomic sequencing is to compare them to a set of labeled reference sequences from a reference database (e.g. RefSeq16 or Virosaurus17 for viruses only). Since these databases are not exhaustive and it is difficult to obtain the full spectrum of genome sequences from diverse, fast evolving organisms (such as RNA viruses18,19), alternative methods are required.
There are implementations of multiple different reference-free (unsupervised) or something in-between (semi-supervised) methods that allow clustering of sequencing reads.20 One can either cluster sequencing reads directly (assembly free) or create longer contigs first by using assembly methods that are specifically optimised for metagenome datasets21.
The advantage of creating longer contigs first, especially for a short read sequencing dataset, is that there is more information present in such contigs and that tools for gene prediction and genome annotation (e.g. Prokka22 or ABRicate23) can be used to further characterize the organism. The disadvantage or limitation of these assembly methods is that they are less sensitive. In cases where there are only a few sequencing reads of an organism present in the dataset, an assembly method will be unable to create a longer contig. It is also possible that artifacts within the assembly (mix of multiple genomes in one contig) are created. There are tools to assess the quality of the resulting contigs. However, they are often designed for bacterial data as bacterial marker genes or codon usage are used for the assessment. In addition, contigs are also typically filtered by their length. These length thresholds are usually above the length of an average viral genome, as viruses generally have shorter genomes compared to prokaryotes (Figure 2.1 shows the length distribution of all viral genomes/segments from the RefSeq database).
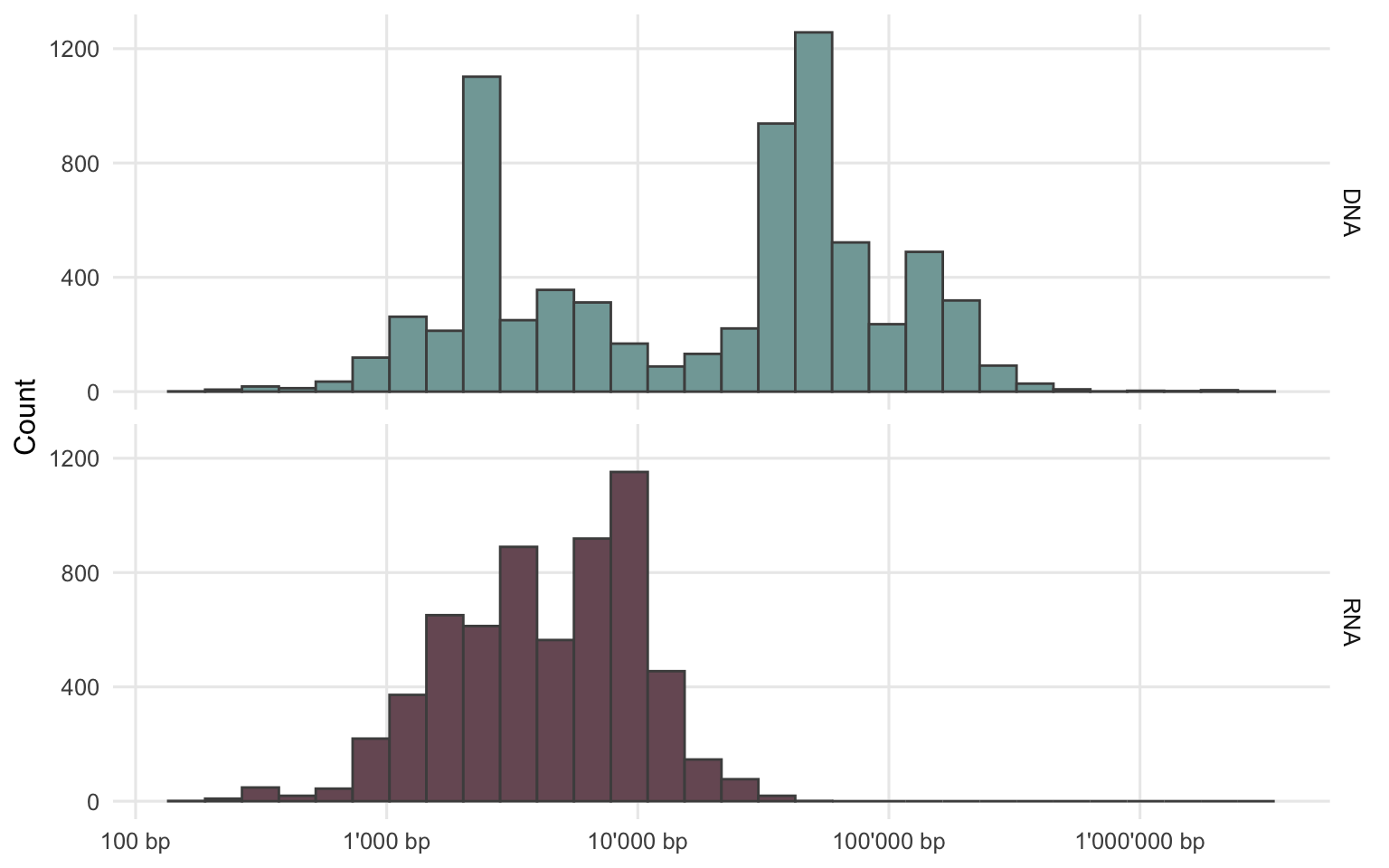
Figure 2.1: Genome or segment length of viruses from the RefSeq database.
References
11. Ma, X. et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biology 20, 50 (2019).
12. Bioinformatics, e. Trimming adapter sequences - is it necessary?
14. Ewing, B. & Green, P. Base-Calling of Automated Sequencer Traces Using Phred. II. Error Probabilities. Genome Research 8, 186–194 (1998).
15. Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
17. Gleizes, A. et al. Virosaurus A Reference to Explore and Capture Virus Genetic Diversity. Viruses 12, (2020).
18. Biek, R., Pybus, O. G., Lloyd-Smith, J. O. & Didelot, X. Measurably evolving pathogens in the genomic era. Trends in ecology & evolution 30, 306–313 (2015).
19. Peck, K. M. & Lauring, A. S. Complexities of Viral Mutation Rates. Journal of Virology 92, (2018).
20. Pérez-Cobas, A. E., Gomez-Valero, L. & Buchrieser, C. Metagenomic approaches in microbial ecology: An update on whole-genome and marker gene sequencing analyses. Microbial Genomics 6, (2020).
21. Ayling, M., Clark, M. D. & Leggett, R. M. New approaches for metagenome assembly with short reads. Briefings in Bioinformatics 21, 584–594 (2020).
22. Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics (Oxford, England) 30, 2068–2069 (2014).
23. Seemann, T. Tseemann/abricate. (2021).